Hereditary angioedema (HAE) is a disease characterized by recurrent episodes (also called attacks) of severe swelling of the skin and mucous membranes. The age at which attacks begin varies, but most people have their first one in childhood or adolescence. The frequency of attacks usually increases after puberty. Attacks most often affect 3 parts of the body:
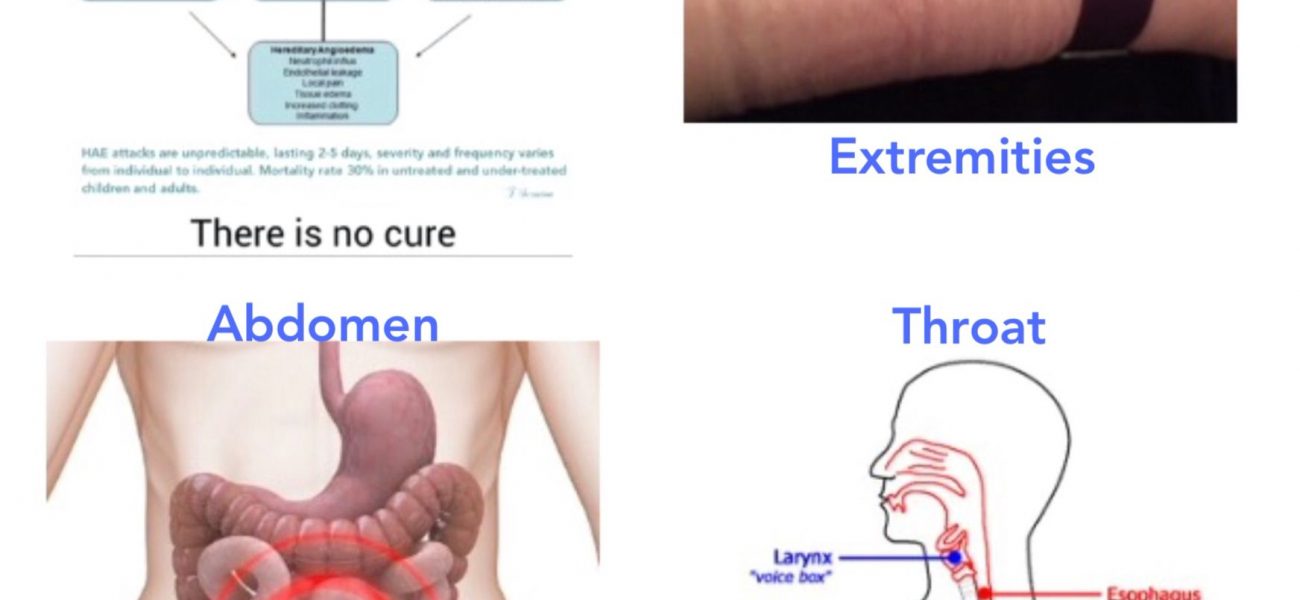
Skin – the most common sites are the face (such as the lips and eyes), hands, arms, legs, genitals, and buttocks. Skin swelling can cause pain, dysfunction, and disfigurement, although it is generally not dangerous and is temporary.
Gastrointestinal tract – the stomach, intestines, bladder, and/or urethra may be involved. This may cause symptoms such as nausea, vomiting, diarrhea, and abdominal pain.
Upper airway (such as the larynx and tongue) – this can cause upper airway obstruction and may be life-threatening. The majority of attacks affecting the airway resolve before complete airway obstruction.
Attacks may involve one area of the body at a time, or they may involve a combination of areas. They always go away on their own but last from 2 to 4 days. While people with HAE have reported various triggers of attacks, emotional stress, physical stress, and dental procedures are the most commonly reported triggers.
There are several types of HAE. Types I and II are caused by mutations in the C1NH gene (also called the SERPING1 gene), which provides instructions for making the C1 inhibitor protein. Type I is due to deficiency of C1 inhibitor, and type II is due to dysfunction of C1 inhibitor. These types are also characterized by abnormal complement protein levels. Inheritance of types I and II is autosomal dominant, but not all people with a SERPING1 gene mutation will develop symptoms of HAE. A third type is called HAE with normal C1 inhibitor. This type is characterized by normal C1 inhibitor and normal complement protein levels, and usually begins in adulthood. While some cases of type III are due to mutations in the F12 gene, in other cases the cause is not yet known. The inheritance of this form is also thought to be autosomal dominant.
Management of HAE involves treatment of sudden (acute) attacks and preventing attacks (prophylaxis). Treatment for acute attacks in types I and II includes replacement with C1 inhibitor concentrates, a kallikrein inhibitor, or fresh-frozen plasma (by infusion). Sudden attacks involving the upper airway may involve intubation if stridor or signs of respiratory distress are present. HAE with normal C1 inhibitor levels is treated similarly, however C1 inhibitor infusion is not effective. Prophylaxis may involve regular injections of C1 inhibitor concentrates, long-term androgen (male hormone) therapy, or antifibrinolytics.
The long-term outlook varies depending on the frequency and location of attacks, and the severity of attacks in each person. Attacks generally continue throughout life, but the frequency of attacks can be significantly reduced with therapy.
Causes
With this disease, a certain protein in your body is not in balance. This causes tiny blood vessels to push fluid into nearby areas of your body. That leads to sudden swelling.
A problem with a gene that makes a blood protein called C1 inhibitor often causes HAE. In most cases, you don’t have enough of this protein. In others, you have normal levels but it doesn’t work right.
For the most common form of HAE, if one of your parents has HAE, you have a 50% change of having it, too. But sometimes the gene change happens for unknown reasons. If you have the broken gene, you can pass it on to your children.
Symptoms
The main symptom is swelling. You won’t have the itching or hives that people often get with allergic reactions. A bout may last 2 to 5 days.
Treatment
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
- Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
FDA-Approved Treatments
The medication(s) listed below have been approved by the Food and Drug Administration (FDA) as orphan products for treatment of this condition.
- C1 esterase inhibitor (human) (Brand name: Cinryze) – Manufactured by ViroPharma Biologics, Inc.
FDA-approved indication: Approved June 2018 for routine prophylaxis against angioedema attacks in adults, adolescents and pediatric patients (6 years old and above) with Hereditary Angioedema (HAE).
National Library of Medicine Drug Information Portal - Icatibant (Brand name: Firazyr) – Manufactured by Shire Orphan Therapies
FDA-approved indication: Treatment of acute attacks of hereditary angioedema in adults 18 years of age and older
National Library of Medicine Drug Information Portal
Medline Plus Health Information - C1-esterase-inhibitor, human, pasteurized (Brand name: Haegarda) – Manufactured by CSL Behring LLC
FDA-approved indication: For routine prophylaxis to prevent Hereditary Angioedema (HAE) attacks in adolescent and adult patients.
National Library of Medicine Drug Information Portal - Ecallantide (Brand name: Kalbitor) – Manufactured by Dyax Corp
FDA-approved indication: Treatment of acute attacks of hereditary angioedema (HAE) in patients 12 years of age and older
National Library of Medicine Drug Information Portal - C1-esterase inhibitor (recombinant) (Brand name: Ruconest) – Manufactured by Pharming N.V.
FDA-approved indication: Treatment of acute attacks of hereditary angioedema (HAE) in adult and adolescent patients.
National Library of Medicine Drug Information Portal - Lanadelumab (Brand name: Takhzyro) – Manufactured by Dyax Corporation
FDA-approved indication: August 2018, lanadelumab(Takhzyro) was approved as a prophylaxis to prevent attacks of hereditary angioedema (HAE) in patients 12 years and older.
The list of some Hereditary Angioedema medicine: